Capítulo I
Navegadores genómicos
1.1.Conceptos básicos
El genoma de una célula es un repositorio de secuencias de ADN empaquetado en forma de cromosomas. Este material hereditario codifica los genes que una vez descifrados resultan útiles para la síntesis de proteínas y moléculas de ARN no codificante. Junto con estos, cohabitan en el genoma otros elementos que regulan la activación de los genes, proporcionando además una cierta estructuración a la cromatina. Para modelar este complejo escenario biológico dentro de un entorno informático, la secuencia de nucleótidos de cada cromosoma ensamblado del genoma debe transformarse, en primer lugar, en un fichero de texto. Junto con su secuencia, es preciso cartografiar cada cromosoma aportando un segundo tipo de datos denominados anotaciones. Las anotaciones son necesarias para indicar la ubicación exacta de aquellos elementos codificados en una secuencia concreta. El catálogo de elementos biológicos identificables dentro de un genoma está constituido primordialmente por genes, sitios de unión de factores de transcripción, inicios de transcripción, marcas de modificación postraduccional de histonas, regiones repetitivas y polimorfismos.
En función de los nuevos datos aportados por investigaciones más recientes, constantemente ampliamos y mejoramos nuestro conocimiento sobre cualquier función biológica o componente celular. En consecuencia, parece natural pensar que tanto la secuencia como las anotaciones de cualquier genoma necesitan renovarse con cierta frecuencia. Para favorecer la reproducibilidad entre estudios científicos publicados en diferentes momentos, por regla general suele distribuirse la última versión de cada genoma (secuencia y anotaciones) junto con el repositorio de anteriores distribuciones. Cada nueva versión de un genoma establece su propio ensamblado de referencia, posiblemente debido a mejoras en la secuenciación de ese organismo, actualizándose las coordenadas de cada elemento funcional dentro del cromosoma apropiado.
Para comparar correctamente anotaciones existen ficheros de conversión de conjuntos de coordenadas entre distintas versiones del mismo genoma (en inglés, liftover). Es fundamental documentar, por tanto, cuál es la distribución que estamos empleando para facilitar estudios de investigación posteriores. El lector puede encontrar en la Tabla 1 las distribuciones genómicas más recientes de varios organismos modelo. Pese a que no existe un consenso establecido, en el periodo de tiempo en el que un ensamblado está vigente es habitual que aparezcan varias versiones de sus anotaciones.
Una distribución concreta de un genoma viene definida por:
•La secuencia de nucleótidos de cada cromosoma del ensamblado.
•La serie de anotaciones pertenecientes a distintas características que fueron cartografiadas dentro de la secuencia de los cromosomas.
Tabla 1. Discributions actualizadas de los genomas más populares
Cuando analiza la secuencia de una región cromosómica para explorar su contenido, el bioinformático accede a estas informaciones mediante un programa especial denominado navegador genómico (en inglés, genome browser). Generalmente, tanto la secuencia como las anotaciones de todas las distribuciones de un genoma están disponibles de forma gratuita a traves de la Red. Con estos programas, todo el conocimiento aportado por distintos grupos de investigación sobre el genoma está integrado en una sola herramienta, facilitando enormemente el intercambio de información. Los navegadores genómicos proporcionan los datos que poseen sobre cada genoma a través de potentes interfaces gráficos que aumentan su legibilidad. No obstante, estas fotografías del genoma son generadas a partir de simples ficheros de texto que contienen las coordenadas de las anotaciones. Estos archivos suelen distribuirse también de forma separada para realizar tratamientos bioinformáticos de forma local.
Un navegador genómico es una aplicación informática que proporciona un conjunto de herramientas para navegar eficientemente a lo largo de la secuencia de los genomas y de las anotaciones cartografiadas en su interior.
Dada su enorme versatilidad, en un navegador genómico existen varios puntos de entrada para acceder a la información de un elemento en particular. Dependiendo de los datos que conozcamos sobre el objeto de la búsqueda, debemos escoger el modo más adecuado. Resulta frecuente acceder desde varios lugares distintos a la misma información. En la Tabla 2 mostramos algunos ejemplos a la hora de localizar la misma región genómica que contiene un gen de interés para nosotros. Según el método escogido y el nivel de detalle de nuestra búsqueda, es probable que el navegador genómico identifique más de un posible resultado. En ese caso, la exploración visual de las alternativas debería ser suficiente para elegir correctamente la región deseada. Una vez realizada satisfactoriamente la búsqueda, el navegador genómico nos proporcionará una fotografía de las anotaciones disponibles en forma de pistas o carriles (en inglés, tracks). Con este mecanismo de representación, el bioinformático puede comparar fácilmente diferentes elementos anotados sobre una misma región genómica. El navegador genómico proyecta simultáneamente sobre cada pista de su visor genómico los elementos biológicos ubicados en ambas orientaciones de la molécula de ADN.
Tabla 2. Puntos de entrada en un navegador genómico
| Clase | Formato | Ejemplo |
| Región | Coordenadas | chr2:80529003-80531487 |
| Gen | Abreviatura | LRRTM1 |
| Gen | Nombre completo | leucine-rich repeat transmembrane neuronal |
| Gen | Código RefSeq | NM_178839 |
| Proteína | Identificador | LRRTM1 |
| Tránscrito | Identificador | BC045113 |
| Secuencia | FASTA | ATGGATTTCCTGCTGCTCGGTC… |
| Publicación | Referencia | Francks, Maegawa, Mol Psychiatry, 2007 |
Una pista de información contiene la colección de elementos biológicos de una determinada clase disponibles en un navegador genómico para una región específica del genoma. Cada anotación está definida por las coordenadas exactas necesarias para posicionar dicho elemento en el interior de la secuencia y un valor asociado al grado de representatividad de este.
La propia secuencia del genoma es una pista cuyo contenido corresponde a la base observada en cada nucleótido de la región visualizada (cuando el tamaño de esta es inferior a la resolución de la imagen el navegador muestra explícitamente la secuencia de bases). Para gestionar de forma eficiente la proyección de varias pistas simultáneamente, el navegador superpone la información en distintos niveles de datos (ver Figura 1). El usuario puede elegir el elenco de pistas que desea visualizar en cada momento, así como en qué orden desea disponerlas en la ventana gráfica. En tiempo real, el navegador compone una fotografía del genoma en función de nuestras necesidades a partir de los ficheros de anotaciones almacenados en su propia base de datos. El usuario puede configurar esta superposición de pistas, escogiendo entre las diversas pistas disponibles precisamente aquellas que más le interesan. Una vez establecido el contenido del nuevo mapa de anotaciones, el navegador realiza una actualización de la imagen.
Figura 1. Representatión gráfica de anotationes en forma de pistas

Por regla general, las anotaciones están agrupadas en bloques conceptuales biológicamente relacionados. Los bloques de opciones disponibles pueden cambiar en función de la distribución del genoma habilitada. Es posible establecer asimismo el nivel de detalle de cada pista para conseguir representaciones gráficas que faciliten una comparación óptima (desplegar o compactar pistas, ver Figura 1). Existen aplicaciones auxiliares en los propios navegadores genómicos para realizar estas comparaciones de forma cuantitativa, teniendo en cuenta la correlación entre la ubicación de los elementos de diferentes anotaciones.
1.2.Filosofía de la navegación genómica
En la actual era posgenómica los investigadores han desplazado el foco de atención desde las secuencias individuales hacia el análisis a gran escala del genoma. Con el objetivo de poseer una visión más amplia de las anotaciones agrupadas entorno a una región del genoma, aparecen los navegadores genómicos, que integran distintas fuentes de información dispersas por la Red. Estos sistemas redefinen, por tanto, la comunicación con estos complejos repositorios de vastos volúmenes de información. De este modo, navegamos por heterogéneos paisajes genómicos codificados en el interior de los cromosomas, concentrando nuestro interés únicamente sobre determinadas áreas. Desde el genoma completo hasta la secuencia individual de una proteína, el usuario de estos servicios puede recuperar todas las anotaciones con numerosos niveles de detalle. Esta amalgama de recursos también es accesible de forma aislada, menguando sin embargo la efectividad del análisis bioinformático fuera de estos entornos integrados.
Como muestra la Figura 2, a partir de la representación gráfica servida por el navegador genómico podemos visitar las anotaciones particulares suministradas por otro tipo de recursos más específicos. Por ejemplo, cuando analizamos los genes anotados en una región genómica podemos acceder desde la pista individual a una nueva pantalla que contiene información recopilada por múltiples fuentes de información. Muchos de estos repositorios, que denominaremos bases de datos primarias, constituyeron en su momento el germen de los actuales navegadores genómicos. Cuando no era posible llevar a cabo análisis globales de un genoma completo, la unidad de búsqueda de información era precisamente la secuencia individual cuya anotación era suministrada por diferentes miembros de la propia comunidad científica.
Figura 2. Paradigma de la comunicatión con navegadores genómicos

El usuario del navegador puede acceder a múltiples recursos integrados en el mismo interfaz
Estas primeras colecciones de secuencias, a pesar de su enorme valor, contenían frecuentes errores que podían conducir a importantes excesos por redundancia. Ahora estos catálogos reciben un tratamiento de validación mucho más cuidadoso, basado en la verificación manual llevada a cabo por expertos entrenados en este tipo de tareas.
Los navegadores genéricos integran la secuencia y las anotaciones de múltiples genomas, presentando todos los datos, por tanto, dentro de un marco común uniforme que facilita su manipulación. El navegador genómico UCSC o la plataforma ENSEMBL son las aplicaciones genéricas más populares. Todas estas herramientas suelen cruzar sus pistas de anotaciones, facilitando de este modo la integración y visibilidad de diferentes fuentes de información desde cualquier lugar. El código de la mayoría de estas plataformas se distribuye gratuítamente junto con todos sus bancos de datos. Profundizando en esta filosofía, la herramienta Gbrowse ofrece todas las funciones de un navegador genómico genérico adaptable a cualquier genoma, cediendo al usuario la responsabilidad de introducir las anotaciones que deben respetar ciertas pautas sobre el formato de los ficheros.
Para determinadas especies se han diseñado navegadores específicos suministrados por dichos consorcios de secuenciación que permiten acceder exclusivamente a sus anotaciones. Estos recursos están optimizados para trabajar con ese genoma, conviertiéndose en un repositorio de referencia para la comunidad de estudio de dicho organismo. Los ejemplos más conocidos de estos programas están dedicados a distintos organismos modelos como FlyBase para la mosca de la fruta, los navegadores de ratón y rata o los portales para la secuenciación de numerosas especies vegetales. Los navegadores genéricos, no obstante, comparten con estos las mismas distribuciones de los genomas (Figura 3). La comparación de regiones ortólogas en un conjunto de genomas puede proporcionar una información muy valiosa sobre la ubicación de ciertos elementos funcionales conservados evolutivamente. Por ejemplo, la identificación de algunas secuencias preservadas a lo largo de millones de años permite refinar la anotación de genes y elementos asociados a su regulación. VISTA es uno de los portales pioneros en realizar este tipo de comparaciones.
En definitiva, el analista bioinformático posee a su disposición un amplio abánico de recursos genómicos (ver Tablas 3 y 4). A lo largo de este libro analizaremos el funcionamiento de algunos ejemplos de estos repositorios de información biológica de enorme valor en un entorno de investigación.
| Recurso primario | Dirección |
| GenBank | http://www.ncbi.nlm.nih.gov/genbank |
| Gene | http://www.ncbi.nlm.nih.gov/gene |
| RefSeq | http://www.ncbi.nlm.nih.gov/refseq |
| Gene Ontology | http://www.geneontology.org |
| GEO | http://www.ncbi.nlm.nih.gov/geo |
| dbSNP | http://www.ncbi.nlm.nih.gov |
| OMIM | http://www.ncbi.nlm.nih.gov/omim |
| Pubmed | http://www.ncbi.nlm.nih.gov/pubmed |
| Navegador genómico | Dirección |
| UCSC | http://genome.ucsc.edu |
| ENSEMBL | http://www.ensembl.org |
| NCBI | http://www.ncbi.nlm.nih.gov |
| Mouse GID | http://www.informatics.jax.org |
| Rat GD | http://rgd.mcw.edu |
| FlyBase | http://flybase.org |
| WormBase | http://www.wormbase.org |
| Saccharomyces GD | http://www.yeastgenome.org |
| Gbrowse | http://gmod.org/wiki/Gbrowse |
| Navegador genómico avanzado | Dirección |
| VISTA | http://pipeline.lbl.gov |
| rVISTA | http://rvista.dcode.org |
| enhancerVISTA | http://enhancer.lbl.gov |
| ENCODE | http://encodeproj ect.org/ENCODE |
| modENCODE | http://www.modencode.org |
| International Cancer Project | http://www.icgc.org |
| International HapMap Project | http://www.hapmap.org |
| Human Epigenome Consortium | http://www.epigenome.org |
Tabla 4. Selectión de recursos genómicos fundamentales(2)
| Comparación de secuencias | Dirección |
| BLAST | http://blast.ncbi.nlm.nih.gov |
| CLUSTALW | http://www.ebi.ac.uk/Tools/msa/clustalw2 |
| TCOFFEE | http://www.tcoffee.org |
| MAFFT | http://mafft.cbrc.jp/alignment/software |
| MAUVE | http://gel.ahabs.wise.edu/mauve |
| MEME | http://meme.nber.net/meme |
| BOWTIE | http://bowtie-bio.sourceforge.net |
| Predicción computacional de genes | Dirección |
| GENEID | http://genome.erg.es/software/geneid |
| GENSCAN | http://genes.mit.edu/GENSCAN.html |
| FGENESH | http://www.softberry.com |
| N-SCAN | http://mblab. wustl.edu/nscan |
| Anotación de regiones reguladoras | Dirección |
| TRANSFAC | http://www.gene-regulation.com |
| JASPAR | http://jaspar.genereg.net |
| ABS | http://genome.erg.es/datasets/abs2005 |
| OREGANNO | http://www.oreganno.org |
| CBS | http://compfly.bio.ub.es/CBS |
| RSA tools | http://rsat.ulb.ac.be |
| WEBLOGO | http://weblogo.berkeley.edu |
| TF-map alignments | http://genome.erg.es/software/meta |
| Aplicaciones de propósito general | Dirección |
| GALAXY | https://main.g2.bx.psu.edu |
| DAVID | http://david.abcc.ncifcrf.gov |
| BABELOMICS | http://babelomics.bioinfo.cipf.es |
| CIRCOS | http://circos.ca |
| GFF2PS | http://genome.erg.es/software/gfftools |
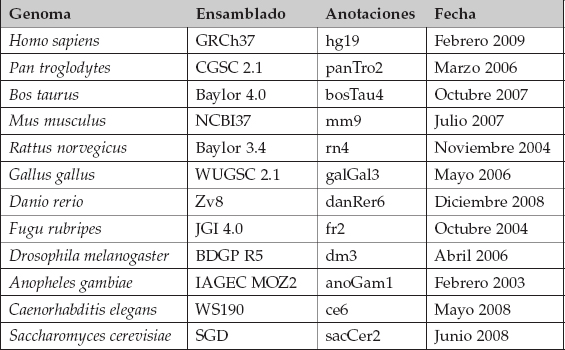
Figura 3. Visualization de una región genómica
El gen lpp visualizado a través de distintos navegadores genómicos. Desde el margen superior de la imagen hacia el inferior podemos ver UCSC, ENSEMBL, Flybase y VISTA.
En otro orden de cosas, el análisis en profundidad del genoma humano resulta de indudable interés dentro del campo de la Biomedicina y la salud, debido a que la mayoría de enfermedades documentadas poseen un importante componente genético. No es casual, por tanto, que sean necesarios también navegadores genómicos que proporcionen un mapeado completo de las anotaciones referentes a estos desórdenes hereditarios. Ejemplos de estos portales biomédicos son el proyecto de secuenciación del cáncer o la detección del conjunto de polimorfismos existentes en el ser humano.
1.3.El navegador genómico UCSC: estructura
El navegador de la Universidad de Santa Cruz de California (UCSC, de ahora en adelante) fue diseñado con el objeto de construir una herramienta que proporcionara al usuario un acceso sencillo a las anotaciones existentes sobre el genoma humano, que estaba siendo secuenciado en aquel momento. Desde su nacimiento, este portal ha incorporado numerosas mejoras en su implementación inicial, conviertiéndose en un recurso de referencia utilizado, por ejemplo, para distribuir los resultados del proyecto ENCODE. Todas las anotaciones sobre un genoma base de referencia se suministran en forma de pistas de datos, gráficamente representadas sobre una línea horizontal en paralelo con la propia secuencia de cada cromosoma. Junto con las pistas servidas por defecto el usuario puede visualizar sus propias pistas dentro del navegador.
Figura 4. Pantella inicial de UCSC

Para iniciar la exploración debemos seleccionar primero el organismo y la versión del genoma que deseamos explorar. A continuación, es necesario introducir la información suficiente para que el navegador genómico pueda identificar el objeto de nuestra búsqueda (p.e. genes, regiones o cromosomas). Vamos a centrarnos en un gen humano denominado LRRTM1 (abreviatura de Leucine-Rich Repeat TransMembrane neuronal 1).
Figura 5. Parámetros de busqueda en UCSC
En función del tipo de identificador especificado, los resultados de la búsqueda serán más o menos concretos, siendo preciso incluir en algunas ocasiones información adicional para caracterizar mejor aquello que deseamos localizar. En la Tabla 5 mostramos distintos ejemplos con diferentes tipos de valores aceptados por el motor de búsqueda del navegador UCSC para acabar encontrando el mismo gen en todos los casos.
Tabla 5. Objectos de búsqueda de UCSC
| Nombre | Ejemplo |
| Cromosoma | chr2 |
| Región | chr2:80529002-80531487 |
| Región | chr2:80529002+2485 |
| Gen | LRRTM1 |
| Tránscrito | NM_178839 |
| Proteína | NP_849161 |
Una vez acotados los parámetros de búsqueda, el sistema procesa estos datos y proporciona un listado de todas las pistas que posean algún nexo en común con la información suministrada. El usuario debe seleccionar uno de estos enlaces para obtener una representación gráfica. En este caso (ver página siguiente), encontramos siete pistas relacionadas con el gen LRRTM1 en la distribución hg19 del genoma humano (a noviembre de 2010).
Figura 6. Listado de pistas para el gen LRRTM1 (resumen)

Para mostrar cómo interpretar esta información procederemos a enumerar los distintos resultados obtenidos, analizando la ficha mostrada en esta misma página:
1.Pista UCSC Genes: dos isoformas de este gen incorporadas dentro de la anotación del genoma humano producida por el propio navegador UCSC.
2.Pista RefSeq Genes: una única anotación producida por el consorcio RefSeq. Este proyecto genera un conjunto depurado de los tránscritos pertenecientes a todos los genes conocidos.
3.Pista Non-human RefSeq Genes: cuatro formas ortólogas de este gen identificadas en las anotaciones servidas por RefSeq para otras especies.
4.Pista ENCODE Gencode: siete isoformas analizadas por ENCODE.
5.Pista Human mRNA: dos tránscritos humanos obtenidos experimentalmente.
6.Pista Non-Human mRNA: dos tránscritos experimentales de otras especies.
7.Pista Vega: siete formas alternativas reconocidas por el consorcio VEGA.
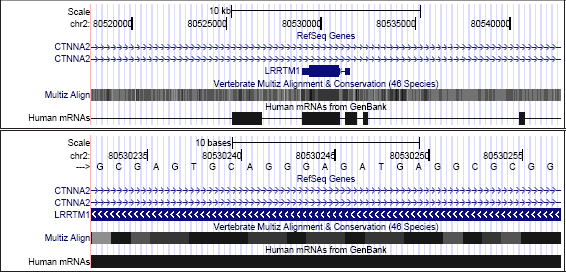
Como ocurre en este ejemplo, suele suceder con cierta frecuencia que para el mismo gen obtenemos anotaciones divergentes producidas por diferentes sistemas de anotación. Para acceder a la representación gráfica del genoma humano que contiene nuestro gen, vamos a seleccionar el enlace asociado a nuestro gen en la pista de RefSeq (RefSeq Genes en la Figura 6), cuyas anotaciones constituyen un estándar de referencia reconocido actualmente por toda la comunidad científica debido a su precisión. Una vez el navegador reconoce nuestra petición, abre la pantalla de navegación principal (ver Figura 7), para automáticamente enviarnos a la región (80,529,003-80,531,487) del cromosoma 2 que contiene este gen según el repositorio de RefSeq. En esta primera conexión el propio navegador UCSC suele acompañar la pista seleccionada con un conjunto predefinido de pistas elementales. El visor genómico está centrado por defecto en la región que contiene exactamente todos los exones de nuestro gen.
Figura 7. Entorno de trabajo de UCSC
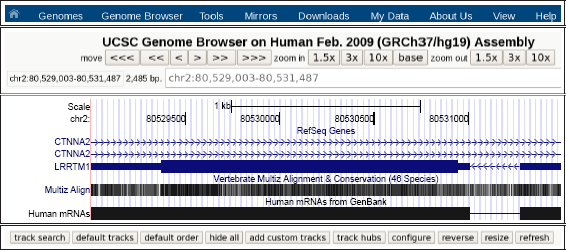
Observamos los dos exones del gen LRRTM1 en forma de cajas rectangulares junto con otras pistas básicas de información (la pista de conservación entre vertebrados y los ARN mensajeros humanos disponibles). Para indicar el sentido de traducción de cada gen, el navegador introduce múltiples flechas a lo largo de los intrones. En este caso, la traducción de este gen debe realizarse de derecha a izquierda (hebra negativa de la molécula de ADN). En ambos extremos del gen, los exones presentan un trazo más fino para distinguir la fracción codificante (CDS, coding sequence en inglés) de la fracción no traducible del tránscrito (UTR, UnTRanslated). Curiosamente, nuestro gen LRRTM1 está ubicado en el interior del intrón de otro gen denominado CTNNA2, codificado en la hebra contraria.
Los navegadores suelen proporcionar un conjunto sencillo de botones de movimiento (Figura 7). UCSC permite realizar movimientos utilizando (a) ampliaciones de posición con diferente resolución para acercar o alejar la escena, (b) desplazamientos horizontales de diferente tamaño a izquierda o derecha de la localización actual, (c) saltos hacia una región concreta mediante la caja de coordenadas y (d) búsquedas rápidas dirigidas por el identificador de cualquier elemento catalogado en este navegador. Una vez accedemos a una pista concreta desde la pantalla inicial, el navegador nos muestra la anotación centrada ocupando toda la ventana gráfica. Para modificar la panorámica podemos alterar la perspectiva usando tres modos de resolución (1.5x, 3x y 10x). A continuación mostramos el efecto de estas funciones sobre la pantalla original:
Figura 8. Jugando con el enfoque de UCSC
Por ejemplo, en ocasiones ocurre que es necesario utilizar zoom out para obtener una visión global del paisaje genómico que rodea a nuestro gen de interés. En otras circunstancias puede ser relevante acceder a fragmentos locales de las anotaciones empleando zoom in. El botón base intensifica al máximo el acercamiento para visualizar directamente la secuencia de nucléotidos. Los botones de desplazamiento (</<</<<< y >/>>/>>>) generan una nueva fotografía de las regiones del genoma inmediatamente anteriores o posteriores a esta ventana. Cada movimiento automáticamente refresca la imagen, actualizando el contenido de las pistas que estemos explorando. Si combinamos la herramienta de ampliación para fijar el ancho de la ventana gráfica con el desplazamiento de esta disponemos de un mecanismo muy efectivo para visitar la sección del cromosoma que más nos interese.
Figura 9. Desplazamiento por el genoma con UCSC

Justo debajo de la imagen generada por el navegador en la Figura 7, el botón reverse permite obtener la anotación gráfica de la región actual seleccionando como referencia de coordenadas la hebra alternativa del mismo cromosoma:
Figura 10. Cambiando la hebra de ADN con UCSC

Como podemos ver en la Figura 10 con esta opción podemos visualizar nuestro gen de interés LRRTM1 mostrando los exones de acuerdo con su propia orientación (obteniendo la ordenación natural de izquierda a derecha, el margen izquierdo de la imagen indica el punto de inicio de la transcripción del gen).
El menú azul superior de la pantalla principal de la Figura 7 contiene enlaces hacia varias aplicaciones complementarias. Dentro del menú denominado View (en inglés, ver), el botón PDF/PS permite realizar una fotografía de la región actualmente visitada, incluyendo todas las pistas activas en ese instante. Esta opción es verdaderamente útil para generar imágenes de alta calidad, que podemos modificar para incluir en nuestras publicaciones científicas:
Figura 11. Fotografiando una region del genoma
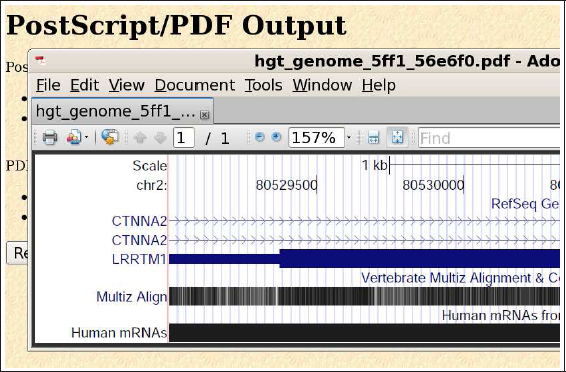
El marco de trabajo establecido por el navegador genómico delimita una serie de acciones que nos permiten modificar dinámicamente la representación gráfica de los elementos anotados en una región genómica. Sin embargo, pese a que la secuencia de nucleótidos no es visible, no debemos olvidar que todas estas anotaciones en forma de pistas contienen información sobre la localización exacta dentro de dicha región de esos elementos.
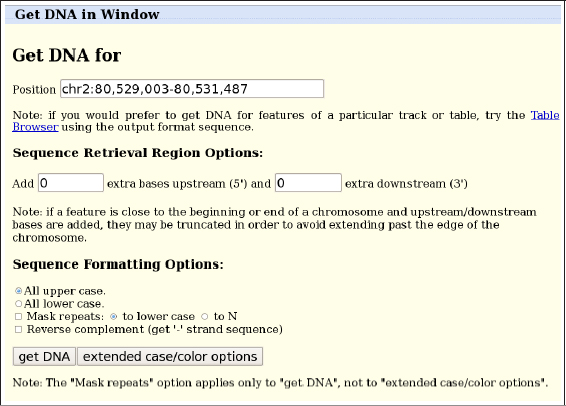
Para obtener directamente la secuencia de nucleótidos fotografiados en la actual ventana, dentro del mismo menú View, podemos utilizar la opción denominada DNA. De este modo accederemos a la secuencia real sobre la cual estamos superponiendo las anotaciones, junto con las regiones flanqueantes a ambos lados de esta:
Figura 12. Extracción de la secuencia de una región genómica
1.4.El navegador genómico UCSC: pistas
Las pistas contienen anotaciones suministradas por distintos consorcios y grupos de investigación. Para mostrar únicamente aquella información relevante en cada caso, el usuario puede configurar dinámicamente el inventario de pistas que desea estudiar, seleccionando el modo más conveniente de visualización para trabajar con cada una durante el análisis. En la parte inferior de la página web podemos encontrar el elenco de las pistas disponibles agrupadas en distintos bloques conceptuales (p.e. genes, regulación, conservación, etc.).
En UCSC podemos modificar simultáneamente el comportamiento de varias pistas, siendo necesario presionar posteriormente el boton refresh (en inglés, refrescar) para reflejar el efecto final de la nueva configuración sobre la región mostrada en el visor. El botón configure permite el acceso a una única pantalla donde podemos modificar de una vez todos los parámetros gráficos del visor genómico.
Para cada distribución de un genoma existe un conjunto determinado de pistas con las anotaciones registradas sobre su secuencia. En consecuencia, el navegador ofrece un conjunto de opciones ligeramente distinto entre diferentes genomas. No obstante, los bloques principales de opciones suelen conservarse dentro de los servicios ofrecidos por el mismo navegador. El listado de bloques proporcionados por UCSC para la versión hg19 del genoma humano puede encontrarse en la Tabla 6. Utilizaremos como referencia esta distribución a lo largo de las explicaciones incluídas en esta sección.
Tabla 6. Bloques de pistas de UCSC
| Nombre | Descripción |
| Mapping and Sequencing Tracks | Ensamblado y secuenciación |
| Phenotype and Disease Associations | Fenotipos y enfermedades |
| Genes and Gene Prediction Tracks | Genes y predicción génica |
| mRNA and EST Tracks | Tránscritos experimentales |
| Expression | Expresión génica |
| Regulation | Regulación génica |
| Comparative Genomics | Genómica comparada |
| Variation and Repeats | Polimorfismos y regiones repetitivas |
Una vez la pantalla del visor genómico de UCSC muestra el inventario de pistas seleccionadas por el usuario, este puede modificar el orden relativo entre las pistas arrastrandolas verticalmente hacia su nueva posición. En función del tipo de elemento biológico, el espacio físico en la pantalla o el tipo de análisis comparativo, en ocasiones debemos modificar la manera en que debe plasmarse gráficamente en nuestro navegador genómico la información sobre las anotaciones. Disponemos de cinco modos de visualización para mostrar una determinada pista sobre la región genómica actual (enumerados de menor a mayor cantidad de información en la siguiente página):
•hide (oculto): no muestra las anotaciones (suele ser la opción por defecto).
•dense (compacto): muestra las anotaciones comprimidas en una sola pista.
•squish (reducido): muestra las anotaciones en varias pistas reducidas.
•pack (agrupado): muestra las anotaciones en varias pistas agrupadas.
•full (completo): muestra toda la información disponible para esa pista.
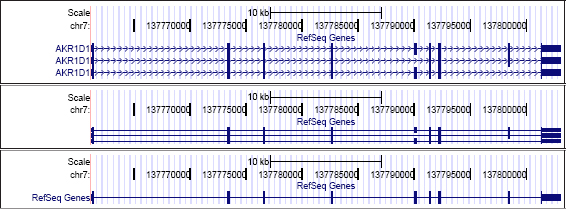
Cada una de estas opciones permite activar gráficamente un conjunto específico de informaciones sobre la pista de trabajo. La visualización de las distintas formas alternativas de un gen es un ejemplo típico que denota la importancia de realizar una selección apropiada de estos datos. A continuación, el lector puede efectivamente verificar cómo podemos plegar y desplegar a nuestro gusto la representación gráfica de las tres isoformas anotadas para el gen humano AKR1D1, según el modo de visualización seleccionado para la pista RefSeq Genes. Primero desplegamos la información básica utilizando pack, después compactamos las tres isoformas cambiando a squish y finalmente plegamos las tres anotaciones en una sola pista con dense:
Figura 13. Configurando la visualizatión de una pista
Para empezar, vamos a proceder a estudiar el bloque de opciones Genes and Gene Prediction Tracks (en inglés, pistas de genes y predicción génica). Este bloque, mostrado en la Figura 14, nos permite acceder a todos los catálogos de genes anotados por diferentes repositorios sobre la región que estamos visualizando en un momento dado. En particular, podemos encontrar la pista de anotaciones RefSeq Genes producida por el consorcio RefSeq.
Figura 14. Bloque de pistas de anotatión de genes de UCSC
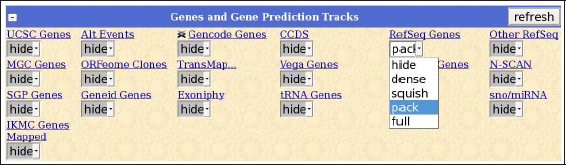
Figura 15. Informatión asociada a la pista RefSeq Genes

Figura 16. Anotaciones para la pista RefSeq Genes

UCSC ofrece información específica sobre cada pista, accesible mediante el enlace coloreado en azul que muestra el nombre de cada pista. A modo de ejemplo, en la anterior página, la Figura 15 muestra, cuando seleccionamos sobre RefSeq Genes en el bloque de opciones de información génica, cómo aparece una nueva pantalla que informa sobre el origen de esas anotaciones.
Para analizar la información asociada a las anotaciones gráficas el usuario debe seleccionar con el ratón sobre el elemento de la pista que desea explorar. En la Figura 7 cuando seleccionamos encima de alguno de los exones del gen LRRTM1 accedemos a cada registro de RefSeq. Podemos ver un resumen del contenido de esta nueva entrada en la Figura 16.
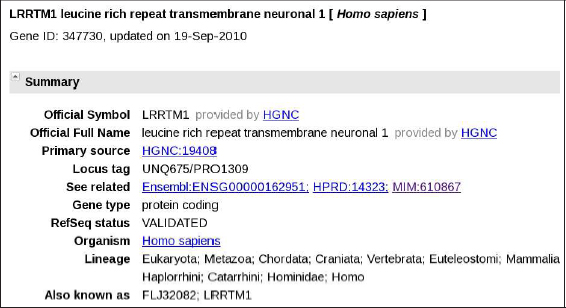
Descendiendo desde la parte superior de la ficha, en primer lugar nos encontramos con el identificador asignado por RefSeq a este gen (NM_178839). A continuación, el navegador nos proporciona los enlaces que ha recopilado para este gen en distintas bases de datos primarias (p.e. Entrez, Pubmed, OMIM, GeneCards, etc.).
Antes de continuar analizando el contenido de la ficha de anotaciones que ofrece RefSeq para nuestro gen dentro del navegador UCSC, proponemos al lector profundizar el recorrido en alguno de estos repositorios. El enlace hacia Entrez Gene (347730), por ejemplo, nos permite acceder a otra ficha de información recopilada por este recurso también gestionado, como RefSeq, por el instituto NCBI (National Center for Biotechnology Information, Centro Nacional para la Información Biotecnológica de los Estados Unidos). En esta nueva página, cada anotación recopilada sobre el gen LRRTM1 posee un enlace que nos permite llegar a la fuente original de esos datos. En la Figura 17 mostramos únicamente el resumen, omitiendo el resto de informaciones. Dentro de esta página, el usuario puede navegar por un amplio conjunto de informaciones, como un gráfico con la estructura exónica del gen, un listado de genes ortólogos en otras especies, un enlace al servidor genómico suministrado por el propio NCBI para navegar por la región colindante o un listado de publicaciones relevantes.
Figura 17. Informatión en Entrez sobre el gen LRRTM1
Aún dentro del documento servido por Entrez Gene, destacamos la anotación de las funciones biológicas que desempeña este gen en la célula por su importancia para caracterizar conjuntos de genes en estudios de análisis masivo de datos genómicos:
Tabla 7. Funciones biológicas del gen LRRTM1
| Función | Evidencia |
| protein binding | computacional |
| axon | bibliográfica |
| NOT cell surface | bibliográfica |
| endoplasmic reticulum | bibliográfica |
| endoplasmic reticulum membrane | computacional |
| growth cone | bibliográfica |
| integral to membrane | computacional |
| membrane | computacional |
Finalmente, desde esta misma entrada de Entrez Gene, el analista bioinformático puede acceder a la secuencia de este tránscrito depositada en GenBank. Este repositorio, pionero durante la década de los ochenta, ofrece información sobre millones de secuencias genómicas y proteómicas. En GenBank, cualquier ficha está estructurada siguiendo un formato notablemente rígido. En cada línea, el tipo de característica anotada se indica primero, mientras que su valor concreto se escribe a continuación. Estas fichas generalmente incluyen información descriptiva y anotaciones sobre una determinada región para acabar con la secuencia completa de esta. Podemos apreciar en la Figura 18 las coordenadas de los dos exones del ARN mensajero del gen LRRTM1 (1..211 y 212..2212) así como su fracción codificante (271..1839). GenBank proporciona también herramientas para manipular las secuencias (p.e. extraer subsecuencias dentro de un cierto rango de coordenadas).
Volvamos a la ficha principal de anotaciones de RefSeq (Figura 16) para estudiar el interfaz denominado Links to sequence (en inglés, enlaces a la secuencia) que permite extraer directamente la secuencia de nucleótidos y aminoácidos de cada gen. Con el enlace Predicted protein (en inglés, proteína predicha) podemos ver el producto de la traducción de este gen, en formato FASTA (Figura 19).
Figura 18. Entra de la secuencia NM_178839 en GenBank

Figura 19. Secuencia de la proteína LRRTM1

Es posible extraer diferentes partes de la secuencia de este gen con la opción Genomic sequence from assembly (en inglés, secuencia genómica del ensamblado). El navegador genómico nos proporciona ahora el acceso a una nueva pantalla donde podemos elegir los componentes de la estructura génica:
Figura 20. Extraer diferntes regiones de un gen con UCSC
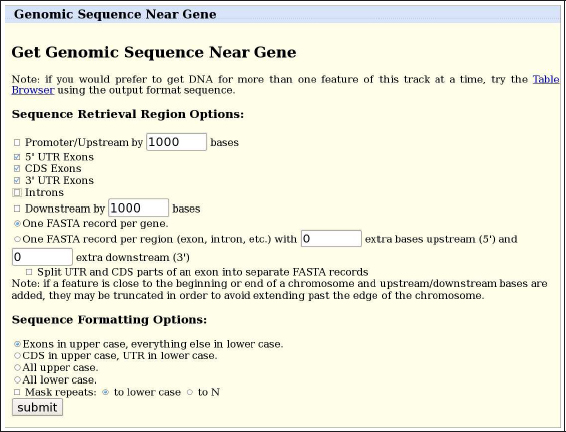
De este modo, es posible obtener un fragmento de la secuencia promotora del gen (Upstream), la región no traducible inicial o final (5’UTR y 3’UTR), la región codificante (CDS) o bien, la secuencia del único intrón de este gen (Introns). Cuando activamos varias opciones simultáneamente podemos combinar las secuencias resultantes que constituyen el propio gen. Cada elemento génico puede separarse en diferentes secuencias en formato FASTA o integrarse en una única secuencia de salida para su posterior análisis.
Una secuencia en formato FASTA está compuesta por un encabezamiento, cuyo primer carácter es el símbolo “>”, que informa de su origen biológico, junto con la propia secuencia a continuación, agrupada en líneas con el mismo número de carácteres.
En la siguiente página mostramos la secuencia completa del gen LRRTM1, presentada en el sentido de lectura original para facilitar la comparación con otros genes anotados en la hebra positiva (ver Figura 21). UCSC realiza automáticamente la operación de complementar las bases e invertir su secuencia para mostrar los datos en cualquier orientación.
Cada exón de este gen está incluido en un fichero FASTA distinto. Utilizamos letras minúsculas para denotar la región no traducible y letras mayúsculas para indicar el fragmento codificante, que empieza con el codón ATG y finaliza con el codón TGA. El navegador UCSC genera suficiente información en el encabezamiento de cada secuencia para una fácil identificación. Podemos reconocer aquí la distribución del genoma (hg19), la pista (refGene equivale a RefSeq Genes), código RefSeq (NM_178839), el exón (0 o 1) y su ubicación genómica.
Junto con los tránscritos humanos revisados manualmente, el proyecto RefSeq produce anotaciones de referencia para varias especies de vertebrados. Esta información resulta extremadamente útil para identificar regiones funcionales conservadas a lo largo de la evolución. Mediante las oportunas comparaciones entre las distintas anotaciones de RefSeq, el propio navegador UCSC habilita una pista adicional dentro del bloque de anotaciones génicas denominada para reconocer las formas ortólogas de cada gen en otros genomas (Figura 22). Con esta información, el usuario debe cambiar el genoma de referencia dentro del navegador y posteriormente utilizar el nuevo código suministrado por RefSeq para recuperar la secuencia homóloga de dicho ARN mensajero.
Figura 21. Secuencia del tránscrito del gen LRRTM1

Figura 22. Formas ortólogas del gen humano KCMF1 en RefSeq
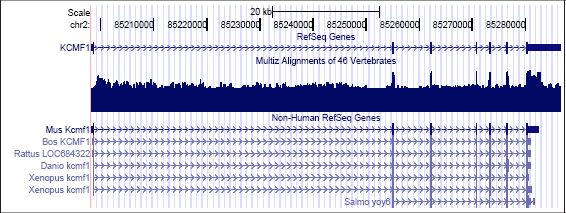
Pueden apreciarse los picos de conservatión sobre las regiones exónicas del gen.
Una secuencia dotada de una función concreta tiende a estar protegida de mutaciones a lo largo de la evolución. En consecuencia, la comparación entre distintos genomas permite revelar aquellos elementos funcionales, que han permanecido invariables. Los bloques de pistas de genómica comparada (en inglés, comparative genomics) permiten realizar múltiples comparaciones entre la secuencia que estamos utilizando de referencia (humano, en este caso, la distribución NCBI36/hg18) y los genomas ensamblados de otros vertebrados:
Figura 23. Bloque de genómica comparada
Figura 24. Conservación de secuencias funcionales entre vertebrados
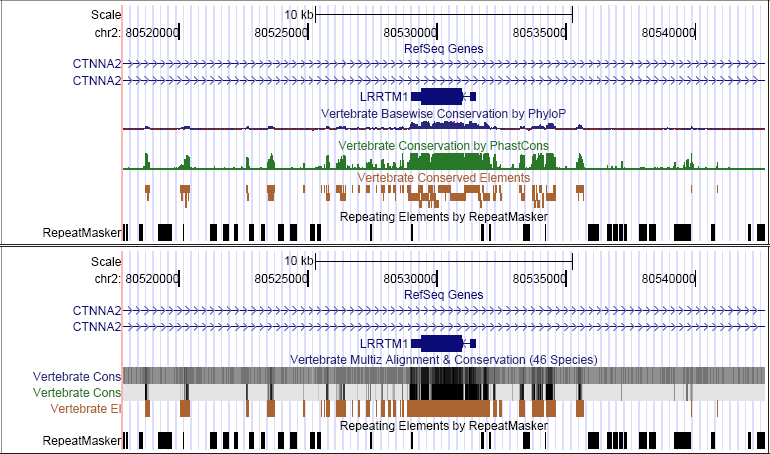
La pista Conservation contiene comparaciones a nivel de secuencia entre la mayoría de las especies más representativas. Para proporcionar estos datos con celeridad, los análisis están precalculados de antemano en el interior del navegador. Con esta pista podemos establecer la existencia de regiones codificantes a través del contraste de las secuencias homólogas a nuestro gen de interés. Esta aproximación también resulta útil para descubrir regiones reguladoras de la transcripción génica. En la Figura 24 mostramos la información sobre conservación para el gen LRRTM1, empleando dos modos de visualización distintos.
Con el navegador UCSC podemos configurar numerosos parámetros de estas pistas para incluir diferentes comparaciones en el resultado final. En este caso concreto, el usuario debe pinchar sobre el enlace coloreado en azul Conservation para acceder a la parametrización de su contenido. En su interior, por ejemplo, tenemos la opción de incorporar o retirar genomas en el visor genómico, modificar el color del gráfico de picos en función de las pautas de lectura o activar diferentes modos de comparación para la identificación de regiones conservadas:
Figura 25. Configuración de la pista Conservation

La herramienta BLAT (accesible desde la barra de menú superior de color azul) complementa perfectamente el bloque de pistas de conservación. BLAT (Blast-like alignment tool) es un programa de alineamiento de secuencias que identifica las regiones más similares a aquella propuesta por el usuario en un determinado genoma. El catálogo completo de genomas servido en el navegador UCSC está preparado para ser interrogado sobre cualquier secuencia de nucleótidos o aminoácidos. Esta aplicación efectúa la búsqueda en las dos orientaciones de la molécula de ADN. A diferencia de otros programas de alineamiento que veremos más adelante, BLAT está especializado en la detección de secuencias extremadamente idénticas. Este hecho aumenta su velocidad de respuesta de forma notable, resultando muy cómodo de utilizar. Podemos emplear esta aplicación para identificar, por ejemplo, cuál es la ubicación del inicio de nuestro gen de trabajo LRRTM1 codificado dentro del propio genoma humano en la hebra (-) de la molécula de ADN:
Figura 26. Funcionamiento de BLAT

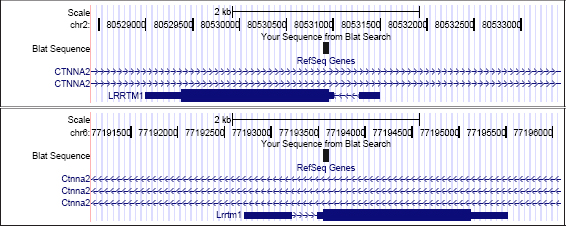
BLAT proporciona una clasificación de posibles ubicaciones de nuestra secuencia ordenadas por similaridad. Aquella opción que seleccionamos como resultado es posteriormente integrada como una nueva pista dentro del navegador genómico. Resulta especialmente interesante el empleo de BLAT para localizar la posición de la región más similar a nuestra secuencia en otro genoma. Con este procedimiento podemos detectar el correspondiente ortólogo del gen humano LRRTM1 anotado dentro del genoma de ratón (ver siguiente página):
Figura 27. Utilizando BLAT para identificar genes ortólogos
1.5.El navegador genómico UCSC: anotaciones propias
El estudio exhaustivo de la cartografía existente para los elementos funcionales codificados en el genoma resulta fundamental para abordar cualquier problema biológico. Sin embargo, cualquier grupo de investigación, durante el proceso de elaboración de hipótesis, habitualmente genera sus propios datos en función de los ensayos llevados a cabo. En consecuencia, es fundamental que el bioinformático conozca los entresijos del manejo de los navegadores genómicos para enriquecer las informaciones originales con los resultados experimentales obtenidos en el laboratorio.
La introducción de anotaciones propias dentro de un navegador genómico resulta esencial para realizar comparaciones entre el conocimiento existente y los nuevos resultados experimentales, favoreciendo tambien la generación de representaciones gráficas de alta calidad.
Una vez el usuario introduce sus propias pistas, estas son automáticamente integradas dentro de la imagen proporcionada por el navegador. Este hecho abre un nuevo abanico de posibilidades, permitiendo el análisis comparativo entre las informaciones existentes y los datos que provienen del laboratorio.
Podemos importar múltiples pistas propias, utilizando el navegador para estudiar el comportamiento de nuestros resultados. De este modo, todas las herramientas de UCSC disponibles para llevar a cabo comparaciones entre distintas anotaciones, tanto a nivel cualitativo como cuantitativo, pueden aprovecharse directamente para contrastar el conocimiento de referencia con nuevos datos experimentales observados para cualquier característica genómica.
Dentro del entorno de trabajo del navegador UCSC, las pistas introducidas en el sistema por el usuario reciben el nombre de custom tracks (en inglés, pistas adaptadas). Desde la pantalla principal del visor genómico, el usuario debe presionar el botón Add custom tracks (añadir pistas) para incorporar una nueva pista de datos. Se permite la carga directa de ficheros comprimidos, lo que incrementa sustancialmente la velocidad de transferencia de los datos a través de la Red. El navegador UCSC asocia un código interno a la conexión realizada desde nuestro ordenador, gestionando internamente las pistas estándar que estamos visualizando y el conjunto de pistas propias que hemos cargado en el sistema. De este modo, estos resultados únicamente pueden ser visibles desde nuestro ordenador, no siendo accesibles para el resto de la comunidad. Por regla general, el navegador mantiene en memoria nuestras pistas durante uno o dos días únicamente, para no almacenar un volumen excesivo de datos.
Para evitar la pérdida de datos, UCSC ofrece un sistema de sesiones de usuario protegidas bajo contraseña (My Data→sessions). Esto permite que el analista bioinformático únicamente cargue sus propias pistas en una ocasión, configurando el resto de pistas de UCSC a su gusto, para grabar ese entorno de trabajo bajo un nombre de sesión. Más adelante, este podrá recuperar la configuración completa rápidamente empleando el enlace obtenido anteriormente sin necesidad de importar más datos. Si el bioinformatico lo considera oportuno, estas sesiones pueden compartirse con otros usuarios, facilitando la comunicación entre diversos colaboradores integrados en el mismo proyecto de investigación que podrán acceder a los mismos resultados mediante la Red.
El bioinformático puede crear sus propias pistas empleando numerosos formatos dentro de UCSC. En esta sección vamos a focalizar nuestro interés en los formatos más populares para la integración de anotaciones genómicas propias: BED, GFF, WIG y BEDGRAPH. Independientemente del formato utilizado para representar los datos en nuestras pistas, UCSC preprocesa estos ficheros para verificar su corrección. En caso de detectar un error de sintaxis, el navegador nos informa para subsanarlo rápidamente.
Nuestras pistas deben poseer la siguiente estructura:
•Configuración del navegador: listado de pistas activas.
•Configuración de la pista adaptada: paramétros de visualización.
•Anotaciones de la pista adaptada: valores de los datos.
En primer lugar, el usuario debe configurar el comportamiento inicial del navegador con la palabra clave browser. Para ello, es necesario introducir las coordenadas de la región genómica que aparecerá al cargar la pista e indicar el conjunto de pistas convencionales que deseamos activar simultáneamente. Es posible seleccionar distintos modos de visualización para estas informaciones:
Figura 28. Encabezamiento de pistas propias (1)

A continuación, dentro del mismo fichero, introduciendo la palabra clave track debemos definir los parámetros de visualización de nuestra pista: asignar un nombre y una descripción, definir un modo de visibilidad y seleccionar un color en formato RGB (Red, Green and Blue, rojo, verde y azul, respectivamente):
Figura 29. Encabezamiento de pistas propias (2)

Finalmente debemos aportar las coordenadas de nuestras anotaciones genómicas para integrarlas en la visualización final. Es fundamental que estas posiciones correspondan a anotaciones mapadas en la misma distribución del genoma que estamos manipulando. Cada línea de nuestras anotaciones deberá reflejar exclusivamente una anotación efectuada por el usuario, introduciéndose el carácter tabulador “ \t ” para separar los distintos atributos de dicho registro.
El formato que debemos emplear para importar información depende de las características de los elementos genómicos incluidos en nuestra pista. Para anotaciones delimitadas necesariamente por una posición inicial y otra posición final es preciso utilizar el formato BED o el estándar GFF, mientras que para mostrar el comportamiento de una determinada característica biológica a lo largo del genoma debemos especificarlo con los formatos WIG o BEDGRAPH.
El formato BED (Browser Extensible Format, del inglés formato extensible del navegador) es útil para plasmar en pantalla aquellas anotaciones constituidas por elementos genómicos definidos por un par de coordenadas (p.e. tránscritos, exones o sitios de unión a factores de transcripción). En su forma más simple, podemos codificar anotaciones proporcionando su localización en el genoma. Para añadir nuevos atributos como el nombre, el color o un valor asociado a su fiabilidad debemos extender la línea actual. Es posible introducir un rango de coordenadas adicional para definir distintos grosores en las cajas (p.e. regiones codificantes y regiones no traducibles de los genes). A continuación, el lector puede observar la sintaxis completa de este formato:
Figura 30. Definición básica y extensiones del formato BED

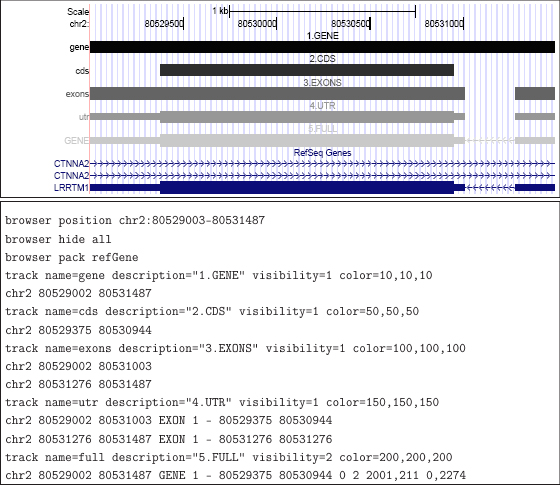
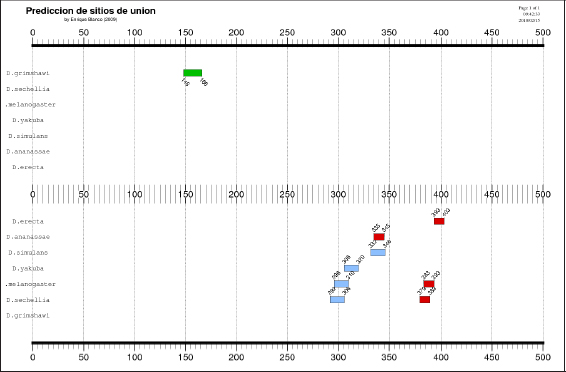
En la Figura 31 hemos ilustrado con un ejemplo real el comportamiento de las anotaciones codificadas en formato BED. Tomando como referencia la información que conocemos sobre la ubicación del gen LRRTM1, analizado a lo largo de este capítulo, hemos generado nuestras propias pistas para resaltar distintas regiones en su interior. A partir de las coordenadas para el propio tránscrito, sus exones y la region codificante, podemos generar tres pistas diferentes (pistas 1, 2 y 3 en la Figura 31), resaltando un área diferente del gen en cada caso. Para denotar con un trazo de distinto grosor la frontera entre la región codificante y la región no traducible, debemos extender el formato BED para añadir una segunda pareja de coordenadas (inicio del CDS, pista 4). Finalmente, para emular la propia representación de la pista RefSeq Genes (con los intrones marcados por una línea recta entre exones y la orientación del gen indicada mediante flechas), enriquecemos el formato añadiendo más información a continuación sobre el número de exones y la longitud de éstos (pista 5, Figura 31).
Figura 31. Anotaciones en distintas versiones del formato BED
El estándar GFF (en inglés General Feature Format, formato de características generales) fue concebido para permitir la transmisión eficiente de anotaciones génicas entre diferentes aplicaciones bioinformáticas. Permite generar representaciones gráficas similares al formato BED, dedicando cada línea a un elemento genómico distinto. Cuando asociamos un identificador propio individualmente a cada anotación, si utilizamos un mismo nombre para un conjunto de anotaciones relacionadas, el navegador interpretará que estas pertenecen al mismo grupo (p.e. los exones de un gen). Es posible asignar una puntuación a cada registro, reflejándose gráficamente con cajas de diferentes grosores o con una gama de colores de distintas intensidades. El formato GFF presenta la siguiente sintaxis básica:
Figura 32. Definición del formato GFF

Existen varias aplicaciones que procesan anotaciones en formato GFF para construir representaciones gráficas de calidad (p.e. los mapas de anotaciones producidas en publicaciones que anuncian la secuenciación de los genomas más populares). Por ejemplo, gracias al programa GFF2PS, las predicciones reportadas por distintas herramientas bioinformáticas pueden volcarse en un único fichero en formato PostScript. En esta representación gráfica, el eje central informa si las predicciones se encuentran en la hebra positiva (arriba) o negativa de la molécula de ADN (abajo):
Figura 33. Representación gráfica de predicciones con GFF2PS
Para personalizar la representación gráfica, el programa GFF2PS permite introducir un fichero de configuración de colores. Ver el manual de usuario del programa para más detalles.
El formato WIG (wiggle, en inglés, agitado) es útil para mostrar la distribución continua de una determinada característica biológica que varía en intensidad a lo largo de un cromosoma (p.e. conservación evolutiva entre especies, muestras de secuenciación masiva, etc.). Para codificarla, el bioinformático debe asignar un valor numérico a cada posición del cromosoma en uso, lo que representa la intensidad de esa señal biológica. Será imprescindible declarar en la cabecera de estas pistas que estamos empleando el formato WIG:
Figura 34. Anotaciones en formato WIG (1)
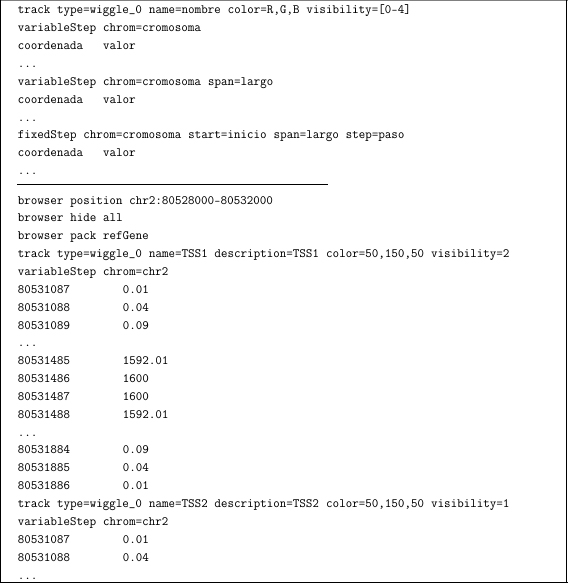
Podemos instruir al navegador para reducir el volumen de información de nuestros ficheros en función de la regularidad de la señal de información. Cuando los datos están ubicados a intervalos regulares de distancia debemos introducir la instrucción fixedStep para no especificar cada posición de la región. Por el contrario, la palabra clave variableStep resulta útil cuando la distancia física entre dos anotaciones consecutivas de nuestra pista no es necesariamente la misma en todos los casos. Además, si un cierto rango de posiciones contiguas presenta el mismo valor, es posible resumir varias anotaciones en una única línea mediante la etiqueta span. A continuación, procedemos a visualizar la pista especificada en la Figura 34 para anotar una hipotética función de probabilidad asociada a la localización del inicio de transcripción de nuestro gen LRRTM1 (mostramos las mismas anotaciones con dos modos distintos de visualización):
Figura 35. Anotaciones en formato WIG (2)

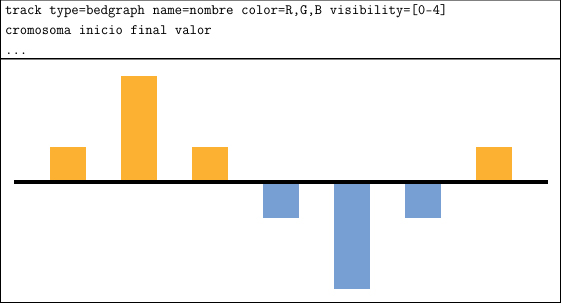
Pese a estos últimos parámetros, los archivos generados bajo el formato WIG poseen un tamaño de varios gigabytes. Para evitar este problema, el formato BEDGRAPH, nacido a partir del formato BED, permite generar representaciones similares con ficheros más reducidos. Con este nuevo formato, es posible aprovechar cada región delimitada por dos coordenadas para dotarla de una altura proporcional al valor promedio de la señal biológica dentro de dicha región. Si suministramos al navegador un conjunto de estas regiones, no necesariamente consecutivas, éste produce un gráfico con nuestros fragmentos intercalados entre zonas sin valor, generando una imagen de valor similar a la producida con el formato WIG, pero reduciendo sustancialmente la cantidad de información que debemos transmitir. Con el formato BEDGRAPH es posible generar fácilmente distribuciones con contraste entre los valores positivos y negativos de la señal:
Figura 36. Anotaciones en formato BEDGRAPH
1.6.El navegador genómico UCSC: ENCODE
La publicación de la secuencia del genoma humano en el año 2000 constituyó un hito de la máxima relevancia dentro de la historia de la Biología y la Medicina. No obstante, gracias a los avances tecnológicos introducidos en la secuenciación durante la última década, progresivamente ha sido posible generar datos de superior calidad con un menor coste económico. En 2004, un consorcio internacional de investigadores iniciaron el proyecto ENCODE (Encyclopedia of DNA Elements, en inglés enciclopedia de elementos del ADN), con el objetivo de identificar con precisión el conjunto de elementos genómicos activados específicamente en el genoma de distintas líneas celulares obtenidas a partir de un amplio catálogo de tejidos. Para lograrlo, el mismo consorcio centró una parte sustancial de sus recursos en el desarrollo de nuevas técnicas experimentales y computacionales que permitieron progresar espectacularmente en múltiples áreas de conocimiento. El navegador genómico UCSC fue la herramienta escogida para centralizar todos estos datos. Las pistas de datos ENCODE se depositaban en UCSC inmediatamente después de llevar a cabo su anotación, existiendo un periodo de cuarentena (nueve meses desde su liberación) que impedía utilizarlas antes en otra publicación, para proteger el trabajo de sus autores.
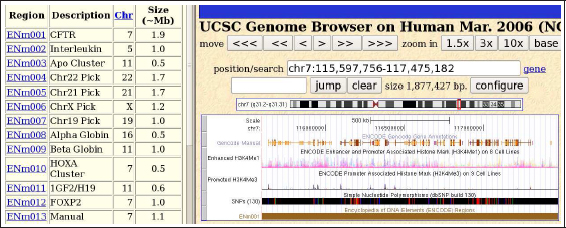
En una primera fase del proyecto, entre los años 2003 y 2007, el consorcio ENCODE dirigió sus esfuerzos sobre el uno por ciento del genoma humano (la fase piloto del proyecto). Para acceder a las anotaciones de esta primera fase, el bioinformático debe seleccionar el enlace ENCODE pilot project en la página principal de UCSC. Una vez allí, puede escoger cualquiera de las cuarenta y cuatro regiones estudiadas para abrir el visor genómico convencional:
Figura 37. Explorando las regiones humanas anotadas en la fase piloto de ENCODE
La anotación definitiva del genoma humano completo fue publicada por el consorcio ENCODE en 2012, contribuyendo sustancialmente a mejorar nuestro conocimiento respecto a los patrones de expresión génica y la regulación de la transcripción en diferentes tejidos. Entre otras líneas de investigación, podemos destacar la elaboración del catálogo de genes de referencia GENCODE mediante la revisión manual de anotaciones automáticas y experimentales, la cuantificación de la expresión génica por secuenciación masiva del transcriptoma de múltiples líneas celulares y la detección de los nucleosomas y las modificaciones postraduccionales de las histonas que configuran la estructura de la cromatina. Para acceder a estos datos debemos pinchar el enlace ENCODE desde la pagina web inicial. La integración de todas estas pistas permite al científico formular cuestiones de indudable relevancia biológica con un nivel de detalle asombroso. Por ejemplo, podemos reconocer qué isoformas de un gen se expresan en un tejido en particular, recuperar el conjunto de polimorfismos anotados sobre sus exones codificantes y estudiar para qué factores de transcripción se ha localizado algún sitio de unión a su región reguladora de la transcripción.
La secuenciación masiva en múltiples líneas celulares de varias marcas postraduccionales de histonas asociadas a activación (H3K4me3 o H3K4me1) y represión génica (H3K27me3) ha generado una gran cantidad de datos. El perfil de ocupación de cada marca a lo largo del genoma presenta un patrón característico de picos asociable a distintos genes. Para gestionar todo este volumen de información los diseñadores de UCSC han creado recientemente un nuevo formato de pista denominado supertrack (en inglés, superpista) que aglutina varias pistas convencionales de datos. Para optimizar el espacio útil en pantalla UCSC proporciona estos datos integrados en una única superpista, que fusiona los patrones observados en todos los tejidos para una marca concreta:
Figura 38. Configuración de la superpista H3K4Mel en ENCODE
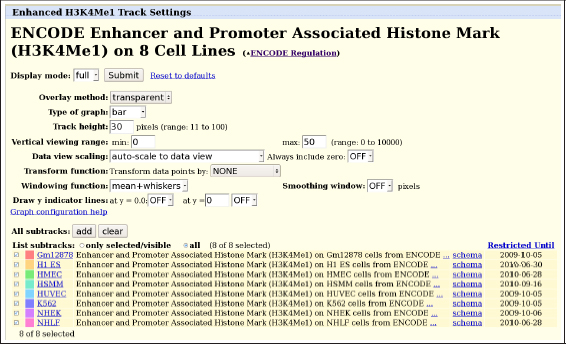
Esta clase de información se integra en una superpista denominada ENCODE Integrated Regulation Track(en inglés, pista de regulacion de ENCODE). Mediante esta superpista podemos manipular simultáneamente datos de secuenciación sobre histonas, factores de transcripción y expresión génica, constituyendo una fuente de información inagotable. A continuación, puede observarse en la Figura 39 la precisión de estas anotaciones para anotar un amplio conjunto de formas alternativas previamente desconocidas de un mismo gen.
Figura 39. Región del genoma humano anotada por el consorcio ENCODE
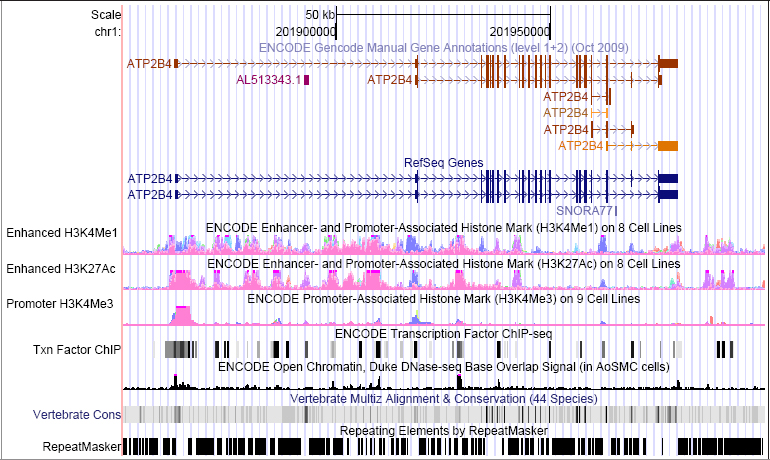
Figura 40.RegiónanotadapormodENCODEenelgenomadeDrosophilamelanogaster
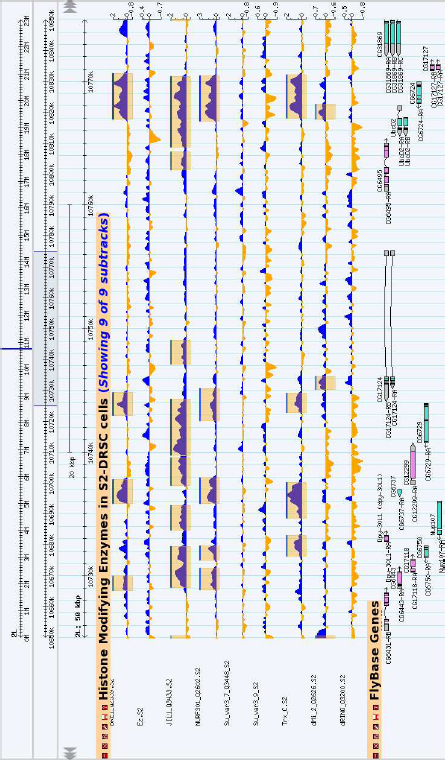
El proyecto modENCODE representa, sin lugar a dudas, una revolución similar dentro de la investigación de dos organismos modelo: la mosca de la fruta Drosophila melanogaster y el gusano de tierra Caenorhabditis elegans. Las anotaciones de este consorcio internacional pueden ser estudiadas mediante el portal dedicado a este proyecto (ver Figura 40).
1.7.El navegador genómico UCSC: navegador de tablas
La herramienta Table browser (en inglés, navegador de tablas), accesible desde el enlace situado dentro de la barra de menú superior de color azul, ofrece al usuario un potente interfaz web para realizar consultas sobre las pistas almacenadas en el navegador de UCSC. En terminología de bases de datos, las pistas reciben la denominación de tablas y cada elemento anotado dentro de una pista es codificado como un registro con una serie de atributos asociados. A diferencia del visor genómico, que representa gráficamente las anotaciones existentes en una región concreta del genoma, el navegador de tablas es un gestor de la base de datos de UCSC que permite interrogar varias pistas simultáneamente para extraer información a gran escala sobre un genoma concreto.
Figura 41. El navegador de tablas de UCSC
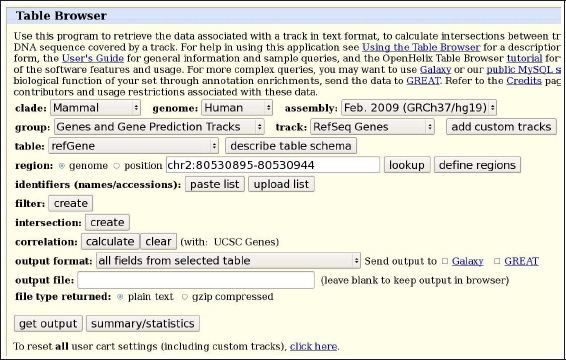
Para trabajar con esta aplicación debemos seguir el siguiente protocolo: (i) seleccionar la distribución del genoma, (ii) establecer la tabla/pista de referencia, (iii) delimitar la región del genoma donde deseamos analizar los datos, (iv) describir los filtros que aplicaremos sobre los registros de la pista/tabla de trabajo, (v) definir las intersecciones con otras tablas/pistas y (vi) seleccionar el formato más apropiado para recuperar los resultados (p.e. secuencias FASTA, listados de datos, pistas integradas dentro del propio navegador).
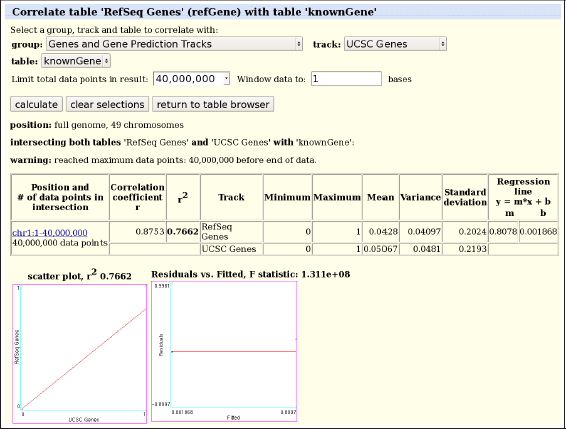
Con esta interfaz es posible efectuar una gran cantidad de cálculos que involucren las anotaciones de varias pistas de UCSC empleando el genoma completo de cualquier especie. Por ejemplo, en el marco del análisis de las regiones reguladoras de un conjunto de genes coexpresados en un experimento a gran escala, podemos extraer las regiones promotoras de los genes implicados en dicho estudio. Como vemos a continuación, también es posible calcular la correlación entre las coordenadas de los genes anotados por RefSeq y la pista :
Figura 42. Cálculo de correlaciones con el navegador de tablas
1.8.El navegador genómico UCSC: instalación local
UCSC suministra anotaciones para más de cincuenta genomas de distintas especies. Pese a que el visor genómico y el navegador de tablas son excelentes herramientas, que conjuntamente ofrecen un amplio abanico de opciones a los usuarios para analizar estas secuencias, en un laboratorio de Bioinformática resulta conveniente obtener una copia local de los genomas para ejecutar protocolos de análisis bioinformático a gran escala1. El enlace Download genomes (en inglés, descargar genomas) permite recuperar los ficheros de texto empleados por el navegador genómico de UCSC para generar las pistas:
Figura 43. Página inicial de descargas de genomas en UCSC

En la Figura 44 ilustramos cómo podemos descargar localmente en nuestro ordenador la información de UCSC. Vamos a focalizar nuestro interés en el genoma humano (hg19), mostrando un esquema de los contenidos de los distintos archivos y directorios.
Inicialmente podemos encontrar los siguientes enlaces en cada distribución (Figura 44A): (i) el conjunto completo de datos de la distribución que incluye los cromosomas y las anotaciones (Full data set), (ii)el conjunto completo de datos de un cromosoma en particular (Data set by chromosome), (iii) las anotacines de cada pista en ficheros individuales (Annotation database), (iv) los ficheros de conversión entre distintas distribuciones (LiftOver files)y (v)las comparaciones con otros genomas (Pairwise alignments/Multiple alignments)
El conjunto de secuencias de nucleótidos de los cromosomas en cada genoma está empaquetado en un único archivo comprimido dentro de la opción Full data set (Figura 44B). Una vez descomprimido este fichero se creará en nuestro ordenador una carpeta con todos los cromosomas separados en diferentes archivos (Figura 44C). Por ejemplo, podemos ver las primeras líneas del cromosoma 17 dentro del fichero chr17. fa presentado en formato FASTA (Figura 44D). También están disponibles las secuencias enmascaradas de cada cromosoma donde se han filtrado las regiones de baja complejidad.
La carpeta (Annotation database) contiene todas las pistas utilizadas por el interfaz gráfico del navegador UCSC (mostramos una pequeña parte de estas en la Figura 44E). En particular, el fichero refGene.txt.gz, que contiene las anotaciones de cada gen humano según el consorcio RefSeq, es procesado por UCSC para generar la pista gráfica Refseq.Genes. Cada línea del fichero refGene.txt codifica la anotación suministrada por RefSeq para un tránscrito humano concreto. En la Figura 44F se muestra la línea correspondiente al único tránscrito del gen LRRTM1. El lector encontrará la descripción de los atributos pertenecientes a este registro en la Tabla 8. Cuando un gen posee varios tránscritos alternativos, estos vienen codificados en líneas diferentes de este archivo.
Recomendamos precaución para procesar las anotaciones de genes codificados en la hebra negativa de la molécula de ADN. En estos casos, las coordenadas iniciales y finales de los registros deben intercambiarse para tener en cuenta este hecho (el campo número cinco representaría entonces el final del gen y el campo número seis el inicio de este). Una conversión similar debería realizarse individualmente con las coordenadas de cada exón a la hora de interpretar su significado para realizar la traducción de los codones correctos.
Figura 44. Descarga de las anotaciones del genoma humano

Tabla 8. Atributos del fichero refGene.txt
| Campo | Ejemplo | Descripción |
| 1 | 1199 | Identificador interno |
| 2 | NM_178839 | Código asignado por RefSeq |
| 3 | chr2 | Cromosoma |
| 4 | - | Hebra |
| 5 | 80529002 | Inicio del tránscrito |
| 6 | 80531487 | Fin del tránscrito |
| 7 | 80529375 | Inicio de la región codificante |
| 8 | 80530944 | Fin de de la región codificante |
| 9 | 2 | Número total de exones |
| 10 | 80529002, 80531276 | Coordenadas iniciales de los exones |
| 11 | 80531003, 80531487 | Coordenadas finales de los exones |
| 12 | 0 | Puntuación |
| 13 | LRRTM1 | Nombre común abreviado del gen |
| 14 | cmpl | Anotación del inicio del CDS (completa) |
| 15 | cmpl | Anotación del fin del CDS (completa) |
| 16 | 0, −1, | Pauta de lectura de los exones |
1.9.El navegador genómico ENSEMBL
ENSEMBL es otro navegador ampliamente extendido que está gestionado por el Instituto Europeo de Bioinformática (EBI) en Hinxton (Reino Unido). En ENSEMBL, las anotaciones se suministran igualmente en forma de pistas que pueden mostrarse o esconderse dentro del visor gráfico. ENSEMBL fue pionero en presentar la información sobre los genes y los tránscritos de forma estructurada en la Red. Los genes, como unidades que agrupan múltiples ARN mensajeros, reciben un identificador con las iniciales ENSG (ENSEMBL gene), mientras que los diferentes productos de este, tránscritos y proteínas, poseen sus propios códigos ENST y ENSP, respectivamente. La plataforma ENSEMBL produce desde sus inicios su propia anotación génica de referencia, colaborando intensamente con varios consorcios para la curación manual de estos datos.
Figura 45. Accediendo a las anotaciones en ENSEMBL
Como se observa en la Figura 45, para acceder a la anotación de un gen el usuario debe seleccionar primero la distribución apropiada del genoma e introducir posteriormente su nombre en la caja de búsqueda.
Figura 46. Anotación del gen LRRTM1 en ENSEMBL
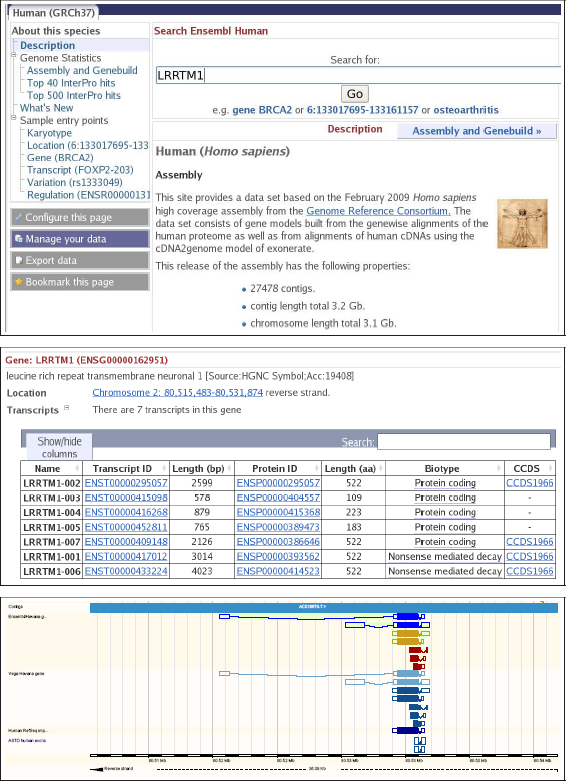
Es posible acceder fácilmente a toda la información recopilada sobre cada gen o visualizar la secuencia de este en pantalla (ver Figura 46). Tanto UCSC como ENSEMBL sirven sus propias anotaciones junto con un conjunto de pistas comunes proporcionadas por otros recursos. La anotación génica de cada navegador puede encontrarse habitualmente como una pista más de datos en el otro programa. Para modificar el inventario de pistas o realizar cambios en su volcado por pantalla, el usuario debe presionar el botón Configure this page (en inglés, configurar esta página). A diferencia del navegador genómico UCSC, que genera imágenes estáticas de las pistas que permiten posteriormente acceder a más información, ENSEMBL reconstruye dinámicamente el visor genómico en el momento de la actualización. A medida que desplazamos el puntero de nuestro ratón sobre ciertas secciones de la imagen, aparece un resumen de la información asociada a cada pista (incluyendo enlaces hacia otras bases de datos). El usuario puede cargar sus propias pistas para integrarlas junto con el resto de información disponible. Con ENSEMBL es igualmente posible generar imágenes para impresión en formato de alta calidad. Los usuarios de ENSEMBL tienen a su disposición numerosos tutoriales, páginas de ayuda e incluso videos con ejemplos ilustrativos (con su propio portal de acceso en YouTube). Para cada opción también existe, específicamente, un manual de asistencia propio.
BIOMART es una de las utilidades más potentes de ENSEMBL. Esta aplicación combina registros de numerosas fuentes con el objetivo de extraer nuevo conocimiento sobre los genes y otras pistas de anotaciones almacenadas en su base de datos. Dado que las acciones que podemos ejecutar con BIOMART o el navegador de tablas de UCSC son ligeramente diferentes, recomendamos que el lector realice practicas intensivas con ambos sistemas. Como se aprecia en la Figura 47, la pantalla de BIOMART está dividida principalmente en dos áreas claramente diferenciadas: el menú (a la izquierda) y la zona de trabajo (ocupando la mayoría de la pantalla). El usuario debe, en primer lugar, establecer un genoma de referencia para trabajar sobre este. La unidad de trabajo en BIOMART es el conjunto de genes de dicho genoma (denominado Dataset en el menú, Figura 47). Inicialmente, podremos realizar operaciones sobre todos los tránscritos anotados por ENSEMBL en el genoma seleccionado (51737 ARN mensajeros en la distribución GRCh37 del genoma humano). Para actualizar la cifra total de genes de trabajo en todo momento, debemos presionar el botón Count tras definir un nuevo filtro sobre los datos.
Figura 47. El interfaz de trabajo de BIOMART
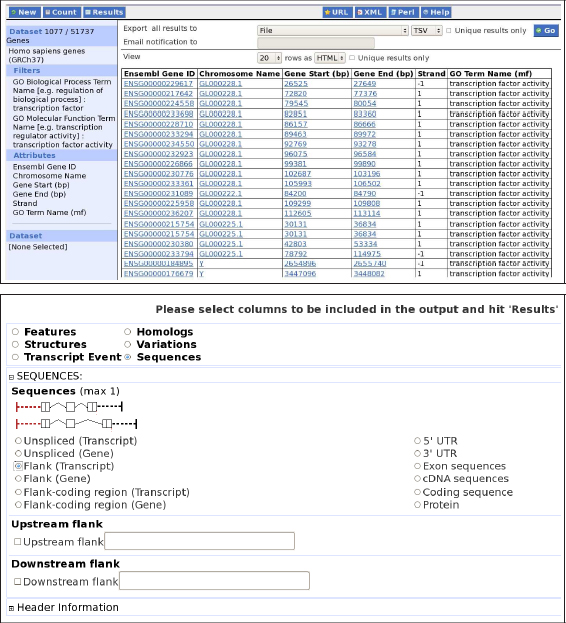
Dado que el contenido de ENSEMBL es actualizado regularmente, es posible que los ejemplos concretos mostrados en esta página no concuerden exactamente con aquellos obtenidos por el lector en el momento presente. En todo caso, el usuario de BIOMART puede optar por trabajar con distribuciones más antiguas de ENSEMBL. Esto resulta especialmente útil para reproducir los resultados comunicados en artículos científicos publicados en años anteriores.
Sobre el catálogo de genes de trabajo realizaremos tres tipos de acciones:
1.Filtros: reducir el conjunto de genes de trabajo, focalizando nuestro interés sobre aquellos que posean determinadas características. Podemos definir filtros sobre cromosomas, regiones genómicas, homología entre especies o funciones biológicas. También es posible introducir nuestra propia lista de genes.
2.Atributos: seleccionar el tipo de información que nos gustaría obtener del conjunto actual de genes. Podemos establecer los atributos generales de cada gen o definir la información sobre homología con otras especies. Existe la opción de extraer sistemáticamente distintas partes de los genes.
3.Dataset: combinar las anotaciones actuales con otro genoma servido por ENSEMBL para reducir el conjunto inicial de resultados.
Cuando hemos finalizado con el filtrado, mantenemos en el área de trabajo precisamente el grupo de genes que cumplen con todos los criterios especificados. En ese instante, justo antes de generar los resultados, es necesario realizar la selección de los atributos. Una vez terminados los procesos de filtrado y formateado de los resultados, debemos utilizar el botón Results para recibir el archivo final. BIOMART exhibe siempre una vista preliminar más reducida de la información solicitada. De este modo, el usuario puede introducir cambios en las preguntas realizadas sobre el catálogo de genes sin necesidad de descargar completamente en cada ocasión el fichero final de resultados (Figura 47).
1.10.La aplicación GALAXY
Es habitual que, para extraer el máximo partido de las anotaciones accesibles con el navegador de tablas de UCSC o BIOMART, sea necesario introducir modificaciones en el formato de esos ficheros. En un laboratorio computacional, estos tratamientos se efectúan localmente en el ordenador del propio analista dentro de un entorno de trabajo gestionado por el sistema operativo LINUX2.
Sin embargo, este tipo de marcos de trabajo, por múltiples causas, están fuera del alcance de los investigadores experimentales. Para facilitar estas modificaciones, el portal web GALAXY ofrece una sencilla implementación de estos mecanismos de análisis. Los ficheros de anotaciones son archivos de texto sin formato, estructurados en filas y columnas, considerándose cada fila un elemento anotado y cada columna un atributo de dicho elemento. De esta manera, el procesamiento de estas estructuras y la comparación con otros conjuntos de datos resulta capital para generar nuevo conocimiento. Como ilustra la Figura 48, GALAXY divide la pantalla de trabajo en tres componentes:
•Menú izquierdo: funciones y servicios ofrecidos.
•Menú derecho: conjuntos de datos y resultados anteriores.
•Pantalla central: resultados y formularios de funciones.
Figura 48. El interfaz de trabajo de GALAXY
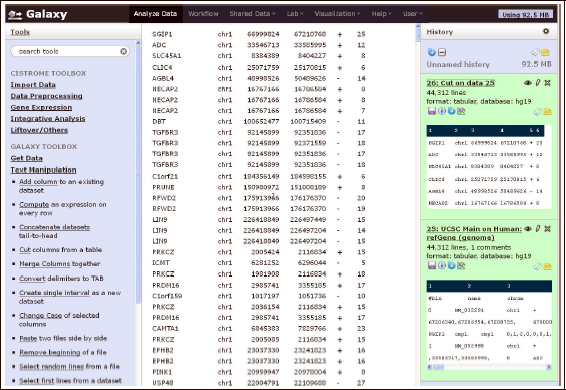
Tabla 9. Opciones de trabajo con GALAXY (selección)
| Bloque | Servicio |
| Conseguir datos | Cargar un fichero de datos del usuario |
| Importar datos del navegador de tablas de UCSC | |
| Importar datos de BIOMART/ENSEMBL | |
| Importar datos del proyecto ENCODE | |
| Importar datos del proyecto modENCODE | |
| Manipulación de texto | Añadir una columna a un conjunto de datos |
| Calcular una expresión sobre cada línea | |
| Fusionar columnas de un conjunto de datos | |
| Crear un intervalo a partir de un rango | |
| Cortar columnas de un conjunto de datos | |
| Elegir al azar algunas líneas de datos | |
| Seleccionar las primeras/últimas líneas | |
| Convertir formatos | De BED hacia GFF |
| De FASTA a formato tabular | |
| De BEDGRAPH a WIG | |
| Manipulación FASTA | Calcular la longitud de una secuencia |
| Filtrar secuencias por longitud | |
| Fusionar secuencias FASTA | |
| Formatear la anchura de las líneas | |
| Convertir secuencias de ADN y ARN | |
| Filtrar y ordenar | Filtrar datos en cualquier columna |
| Seleccionar líneas con una expresión | |
| Ordenar líneas (ascendente/descendente) | |
| Unir, substraer, agrupar | Unir dos conjuntos de datos por una columna |
| Comparar dos conjuntos buscando diferencias | |
| Agrupar datos para eliminar repeticiones | |
| Substraer un conjunto de datos de otro |
La filosofía de trabajo de GALAXY se basa en la sucesiva aplicación de tareas sobre nuestros distintos grupos de informaciones. En primer lugar, el usuario ha de cargar uno o varios ficheros de datos (propios o importados desde un navegador genómico) para comenzar a trabajar. Nuestros conjuntos de datos aparecen siempre en la parte derecha de la pantalla, iniciando nuestro historial de trabajo. Mediante el icono apropiado, podemos visualizar el contenido de estos elementos en la pantalla central. Posteriormente, empleando el amplio abánico de funciones de la parte izquierda de la pantalla para analizar esos conjuntos, el usuario puede realizar múltiples modificaciones sobre dichos conjuntos. En la Tabla 9 hemos seleccionado las opciones de trabajo más habituales. Para ejecutar un servicio, es preciso pinchar en este y una vez en la pantalla central debemos escoger el conjunto datos de nuestro historial que deseamos procesar. También podremos emplear las opciones disponibles para configurar el comportamiento de esta función. Una vez lanzamos el trabajo y este ha finalizado, aparecerá en el historial de la parte derecha un nuevo conjunto de resultados para continuar trabajando. Estos nuevos resultados poseen las mismas propiedades genéricas que el resto de componentes del historial. Las pistas de datos generadas durante nuestros análisis pueden ser descargadas en forma de fichero de texto convencional en cualquier momento.
1Para profundizar en este tema, recomendamos el libro de E. Blanco (2012). Fundamentos de informática en entornos bioinformáticos. Editorial UOC, Barcelona. ISBN: 9788490299982.
2Para más información recomendamos el libro de E. Blanco (2012). Fundamentos de informática en entornos bioinformáticos. Editorial UOC, Barcelona. ISBN: 9788490299982.